Quick start
How do I run CausalCCC ?
This vignette guides you to prepare your Single-Cell data to use CausalCCC. This is a beginner-friendly presentation. See Advanced mode to personalize the CausalCCC pipeline to your biological question.
To create a CausalCCC network, you only need a list of L-R links from your CCC analysis and the raw count expression matrices of the sender genes and the receiver genes you want to study.
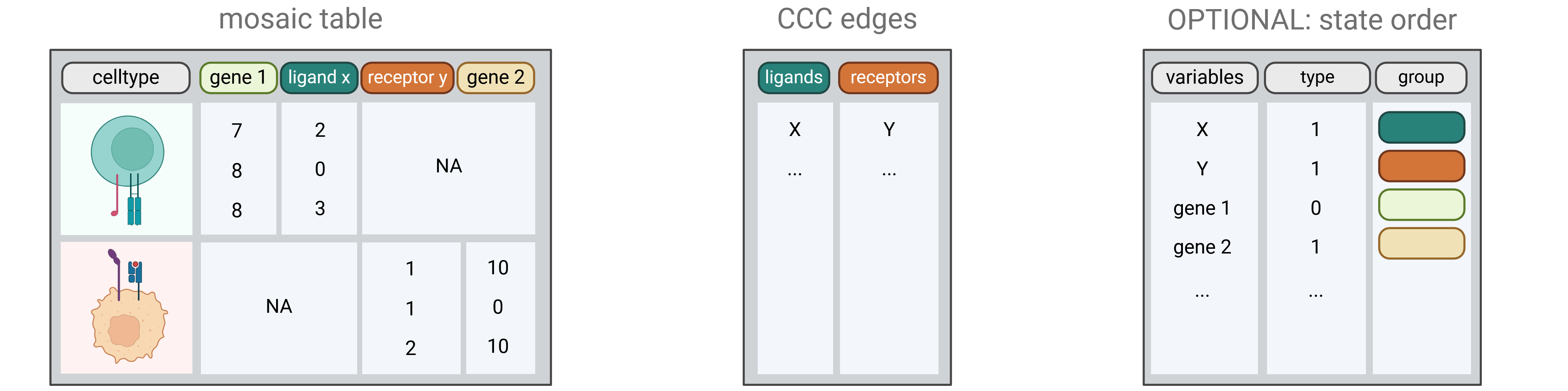
example of causalCCC input files
If needed, we offer in our package:
an optional wrapper function to filter your single-cell data (Seurat, CSV, Excel …).
a feature selection option to help you select genes of interest.
a CCC analysis option to help you select L-R links of interest.
You can perform each or all of these options within one single function call.
my_files <- causalCCC.wrapper(SeuratObject)With this wrapper function you can transform your single-cell object (Seurat example) into CausalCCC input files (wrapper output example).
Preparing your data to submit a causalCCC job
When you have your L-R links, and the genes and cells you want to study, and you just need help formatting your single-cell object.
You can run causalCCC.wrapper() and automatically prepare files to reconstruct an causalCCC network.
Make sure you can load the following packages:
### Mandatory Libraries
library(tidyverse)
library(data.table)
library(dplyr)
### If you use Seurat
library(Seurat)
# You need to download both the MIIC R package adapted for CausalCCC and the CausalCCC package containing preprocessing automatic functions:
Sys.setenv(GITHUB_PAT = 'YOUR_GITHUB_API_TOKEN_HERE')
devtools::install_github("miicTeam/miic_R_package@causalccc", force = T)
devtools::install_github("miicTeam/CausalCCC", force = T)
library(miic)
library(causalCCC)With a Seurat object
#demo dataset
load("causalCCC_demo_Seurat.RData")Download L-R links dataframe example
# Define your senders, your receivers and the metadata where they can be found
senders <- c('B')
receivers <- c('CD8 T')
interact_ident <- "seurat_annotations"
#Define the genes you are interested in reconstructing network (we offer a feature selection tool if needed, see below)
genes_senders <- c("CD74", "RPL11", "RPS2", "RPS15A", "EEF1A1", "RPL10", "GAPDH",
"RPL6", "RPL13A", "RPS14", "RPLP1", "RPS12", "RPL31", "RPS3",
"COTL1", "PTMA", "MALAT1", "RPS4X", "RPS7", "RPL32", "RPS8")
genes_receivers <- c("GZMH", "HIPK1", "PCNA", "BAG2", "NAAA", "ASCL2", "SAMM50",
"CCL4L1", "CD83", "ZNF175", "ACAD9", "SLC12A2", "CD151", "CCL5",
"ATG7", "FABP5", "ZNF438", "NKG7", "FBXO28", "PROK2", "MARS2",
"PRDM15", "NSMCE2", "B2M", "HLA-C", "HLA-A", "HLA-B", "HLA-DRA")
#Load your CCC analysis (we offer a CCC analysis tool if needed, see below)
interact_edges <- read.table("causalCCC_demo_CCClinks.tsv", sep = "\t", header = T)The causalCCC.wrapper() create a folder named “CausalCCC_files” with the wrapper output files in your working directory.
my_files <- causalCCC::causalCCC.wrapper(SeuratObject,
interact_ident = interact_ident,
senders_name = senders,
receivers_name = receivers,
genes_senders = genes_senders,
genes_receivers = genes_receivers,
interact_edges = interact_edges)They are many parameters you can specify for the wrapper function, you can read about them in the R documentation vignette or in Advanced mode.
Now you can run the job on the CausalCCC webserver.
On the workbench you can directly upload the files you just created with the wrapper.
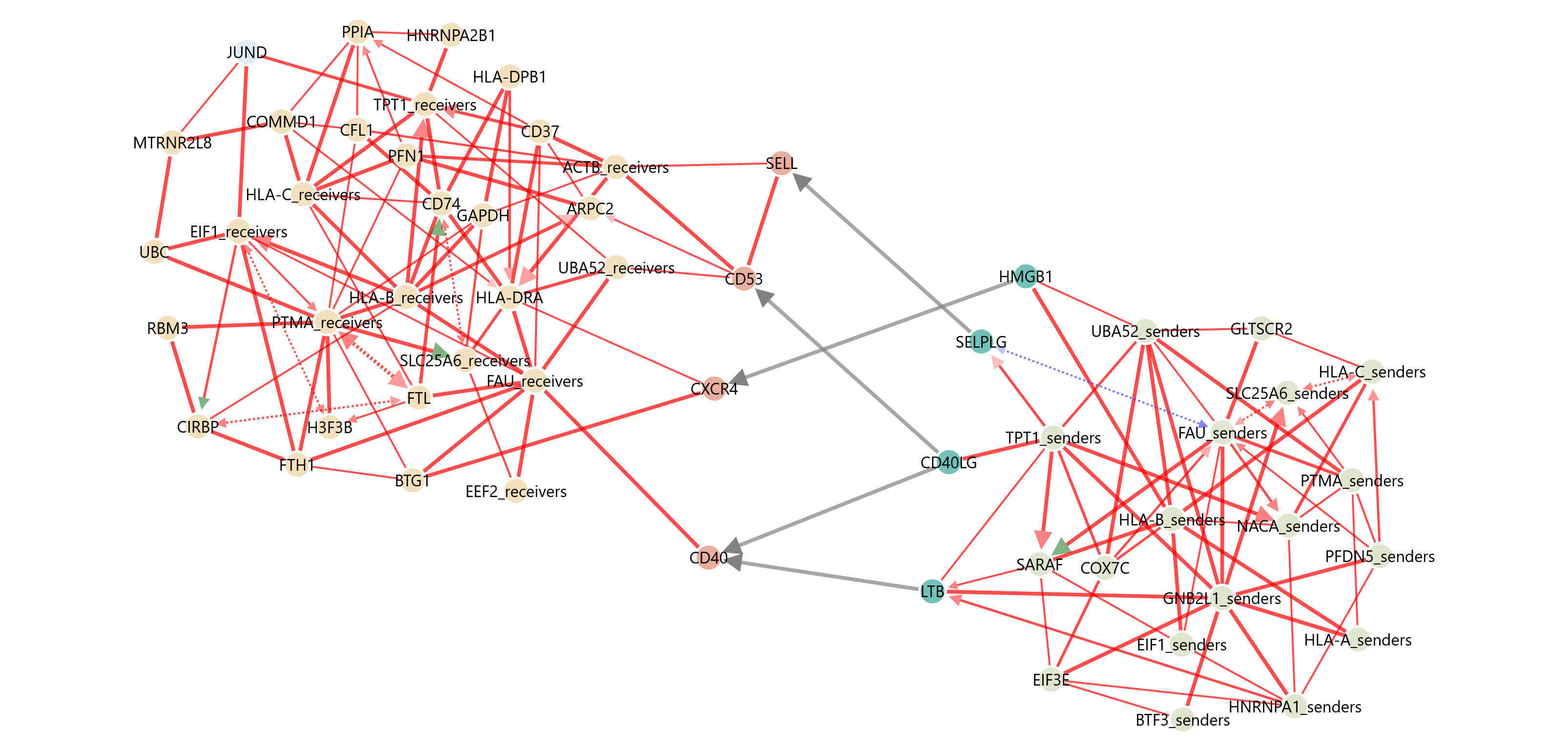
example of the demo causalCCC network on the webserver
With a dataframe
The dataframe can be whichever extension file you like. It must be structured such that columns are genes or metadata and rows are cells.
#demo dataset
load("causalCCC_demo_df.RData")
#or
df <- read.csv("causalCCC_demo_df.csv")
#or
df <- read.csv("causalCCC_demo_table.csv")
#or
df <- read_excel("causalCCC_demo_df.xlsx")Download L-R links dataframe example
# Define your senders, your receivers and the metadata where they can be found
senders <- c('B')
receivers <- c('CD8 T')
interact_ident <- "seurat_annotations"
#Define the genes you are interested in reconstructing network (we offer a feature selection tool if needed, see below)
genes_senders <- c("CD74", "RPL11", "RPS2", "RPS15A", "EEF1A1", "RPL10", "GAPDH",
"RPL6", "RPL13A", "RPS14", "RPLP1", "RPS12", "RPL31", "RPS3",
"COTL1", "PTMA", "MALAT1", "RPS4X", "RPS7", "RPL32", "RPS8")
genes_receivers <- c("GZMH", "HIPK1", "PCNA", "BAG2", "NAAA", "ASCL2", "SAMM50",
"CCL4L1", "CD83", "ZNF175", "ACAD9", "SLC12A2", "CD151", "CCL5",
"ATG7", "FABP5", "ZNF438", "NKG7", "FBXO28", "PROK2", "MARS2",
"PRDM15", "NSMCE2", "B2M", "HLA-C", "HLA-A", "HLA-B", "HLA-DRA")
#Load your CCC analysis (we offer a CCC analysis tool if needed, see below)
interact_edges <- read.table("causalCCC_demo_CCClinks.tsv", sep = "\t", header = T)The causalCCC.wrapper() create a folder named “CausalCCC_files” with the wrapper output files in your working directory.
my_files <- causalCCC.wrapper(df,
interact_ident = interact_ident,
senders_name = senders,
receivers_name = receivers,
genes_senders = genes_senders,
genes_receivers = genes_receivers,
interact_edges = interact_edges)If you need Cell-cell communication analysis
We integrate a CCC analysis using the LIANA pipeline.
### For CCC selection
library(liana)Now you can specify which CCC method you want from LIANA available options :
| Code | Name |
|---|---|
connectome |
Connectome |
logfc |
iTALK inspired 1-vs-rest LogFC score |
natmi |
Network Analysis Toolkit for the Multicellular Interactions |
sca |
SingleCellSignalR |
cellphonedb |
CellPhoneDB |
LIANA is always integrating new CCC methods and this list will be updated as LIANA release new CCC options.
#demo dataset
load("causalCCC_demo_Seurat.RData")Define the parameters as usual, here we show you how to write metadata lists if you want to add one:
# Specify the CCC method you want to use to select L-R edges (optional)
CCC_method <- "natmi"
# Set your working directory here
wd_path <- "~/CausalCCC/vignettes"
# Precise assay of interest
assay_name <- 'RNA'
# Specify the species genes format (mouse or human)
species <- "human"
# Define your senders, your receivers and the metadata where they can be found
senders <- c('B')
receivers <- c('CD8 T')
interact_ident <- "seurat_annotations"
#genes and metadata of interest for the senders population
goi_senders <- c('HLA-DQA1')
metadata_senders <- list()
#genes and metadata of interest for the receivers population
goi_receivers <- c('LAG3')
metadata_receivers <- list()
# exemple:
# metadata <- list(
# treatment = c("Control", "Treated"),
# another_meta = c("Level1", "Level2", "Level3"),
# continuous_meta = NULL
# )By setting do_CCC = TRUE, the wrapper will perform both CCC analysis and feature selection as we assume that without L-R links upstream and downstream genes are undefined.
my_files <- causalCCC.wrapper(SeuratObject,
assay_name = assay_name,
interact_ident = interact_ident,
senders_name = senders,
receivers_name = receivers,
species = species,
do_CCC = TRUE,
CCC_method = "sca",
goi_senders = goi_senders,
goi_receivers = goi_receivers,
metadata_senders = metadata_senders,
metadata_receivers = metadata_receivers,
wd_path = wd_path)If you want feature selection
Maybe you obtained L-R links, but do not know which other genes to include. We offer a unsupervized feature selection tool that will select upstream and downstream genes based on the list of ligands and receptors of interest. Optionally, you may also extend the selection based on a small list of genes and metadata (eg experimental condition, treatment vs control, etc) interesting for your biological question.
By setting do_MIselect = TRUE, the wrapper will chose informative genes based on the variables of interest using mutual information computation.
We showcase it here with a Seurat object but it can be done with dataframes too.
#demo dataset
load("causalCCC_demo_Seurat.RData")Download L-R links dataframe example
# Set your working directory here
wd_path <- "~/CausalCCC/vignettes/"
# Precise assay of interest
assay_name <- 'RNA'
# Specify the species genes format (mouse or human)
species <- "human"
# Define your senders, your receivers and the metadata where they can be found
senders <- c('B')
receivers <- c('CD8 T')
interact_ident <- "seurat_annotations"
#Define optional variables of interest
goi_senders <- c("CD74")
goi_receivers <- NULL
#Load your CCC analysis (we offer a CCC analysis tool if needed, see below)
interact_edges <- read.table("causalCCC_demo_CCClinks.tsv", sep = "\t", header = T)my_files <- causalCCC.wrapper(SeuratObject,
assay_name = assay_name,
interact_ident = interact_ident,
senders_name = senders,
receivers_name = receivers,
species = species,
interact_edges = interact_edges,
do_MIselect = TRUE,
goi_senders = goi_senders,
wd_path = wd_path)